| NAD metabolism is essential for maintaining mitochondrial function and is closely linked to cancer cell survival and therapeutic response. Disruption of NAD homeostasis impairs immune cell function and affects tumor behavior. Recent studies show that age-associated upregulation of CD38 in T cells causes NAD depletion, leading to mitochondrial dysfunction and reduced antitumor activity of CAR-T cells. In clear cell renal cell carcinoma, the mode of NAD⁺ regeneration differs by tumor stage. Primary tumors show low dependence on mitochondrial complex I, whereas metastatic lesions require complex I–mediated NAD⁺ regeneration for survival. These findings indicate that NAD metabolism is a key factor of cancer cell survival and treatment response. |
|
Age-associated nicotinamide adenine dinucleotide decline drives CAR-T cell failure (Nature Cancer, 2025)
Summary: Age-associated upregulation of CD38 in T cells leads to NAD depletion, which causes mitochondrial dysfunction and reduces the proliferative capacity and persistence required for effective CAR-T cell therapy. NMN, a precursor of NAD, is important for maintaining CAR-T cell function. However, in aged T cells, NMN alone is not sufficient, and only the combination of CD38 inhibition and NMN supplementation restores NAD levels, rescues the persistence and antitumor efficacy of aged CAR-T cells.
Highlighted technique: In this study, intracellular NAD levels and the NAD/NADH ratio were measured in CD8 T cells and CAR-T cells from young and aged mice using a NAD/NADH assay, revealing a significant NAD decline in aged T cells. After treatment with NMN alone or in combination with a CD38 inhibitor, the same assay was performed and combined with mitochondrial membrane potential and OCR measurements.
|
|
Mitochondrial complex I promotes kidney cancer metastasis (Nature, 2024)
Summary: Clear cell renal cell carcinoma (ccRCC) shows suppressed mitochondrial and oxidative metabolism during primary tumor growth in the kidney, whereas metastatic ccRCC exhibits a strong dependence on mitochondria. Notably, mitochondrial complex I–mediated NAD⁺ regeneration is required for efficient metastatic survival, while complex I activity is largely dispensable for primary tumor growth.
Highlighted technique: To functionally validate metabolic alterations observed in ccRCC patients, fresh mitochondria were isolated from human primary ccRCC tumors and matched normal kidney tissue, and substrate-defined high-resolution respirometry was used to measure complex I-, II-, and IV–dependent oxygen consumption rates. This analysis directly demonstrated globally impaired mitochondrial respiration, including reduced complex I activity, in primary ccRCC tumors.
|
Application Note I
> Inhibition of Mitochondrial Electron Transport Chain
|
|
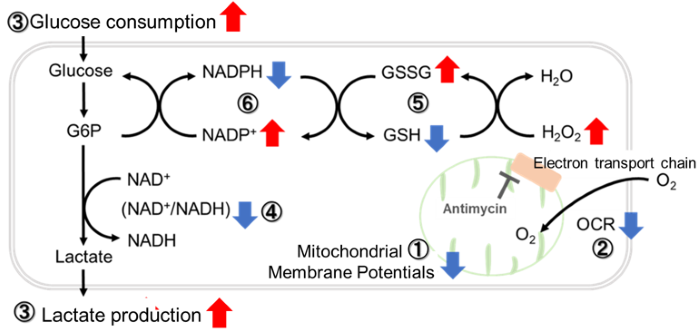
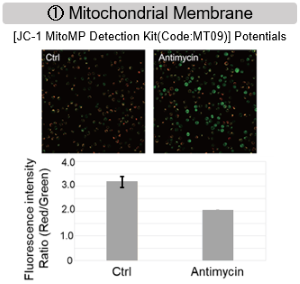
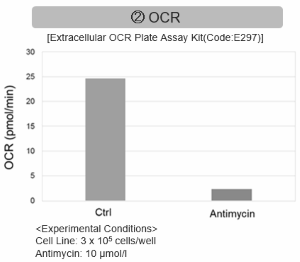
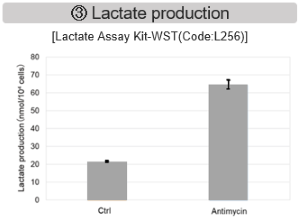
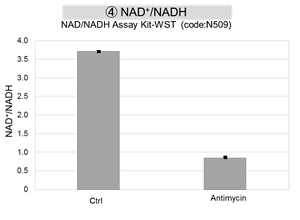
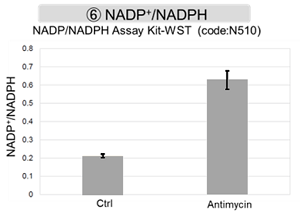
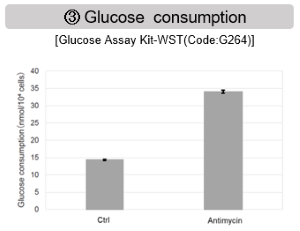
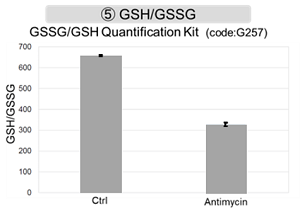
Antimycin stimulation of Jurkat cells was used to evaluate the changes in cellular state upon inhibition of the mitochondrial electron transport chain using a variety of indicators.
The results showed that inhibition of the electron transport chain resulted in (1) a decrease in mitochondrial membrane potential and (2) a decrease in OCR. In addition, (3) the NAD+/NADH ratio of the entire glycolytic pathway decreased due to increased metabolism of pyruvate to lactate to maintain the glycolytic pathway, (4) GSH depletion due to increased reactive oxygen species (ROS), and (6) increase in the NADP+/NADPH ratio due to decreased NADH required for glutathione biosynthesis were observed.
|
|
Application Note II
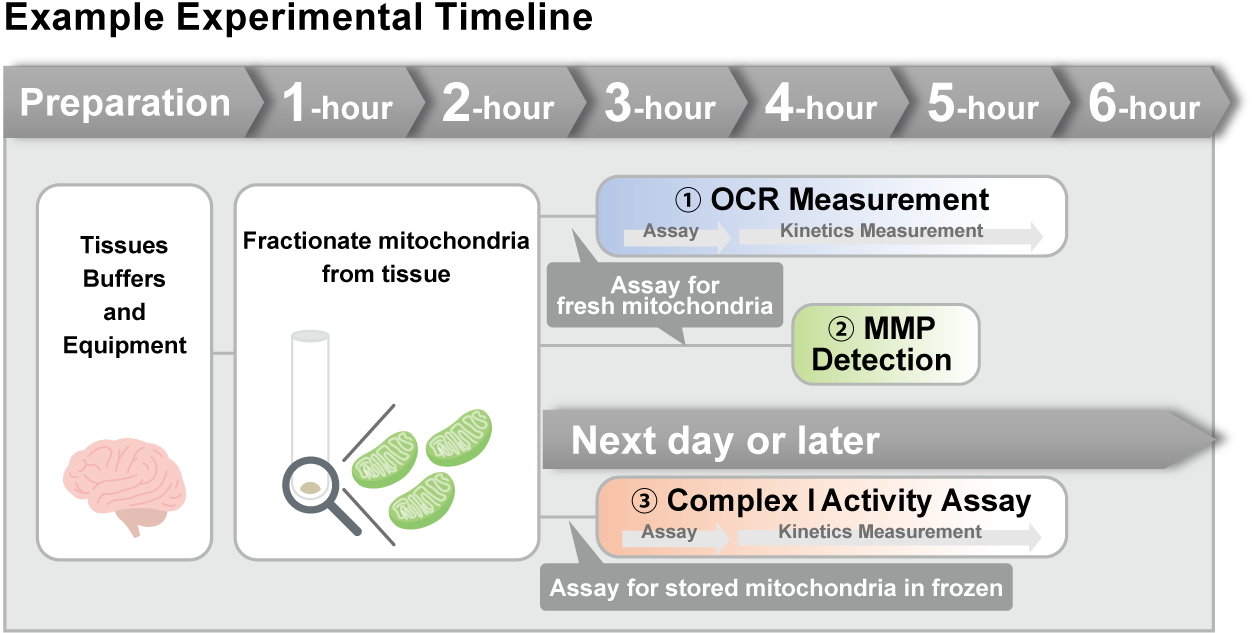
> Activity Evaluation of Mitochondria Fractionated from Mouse Brain
|
|
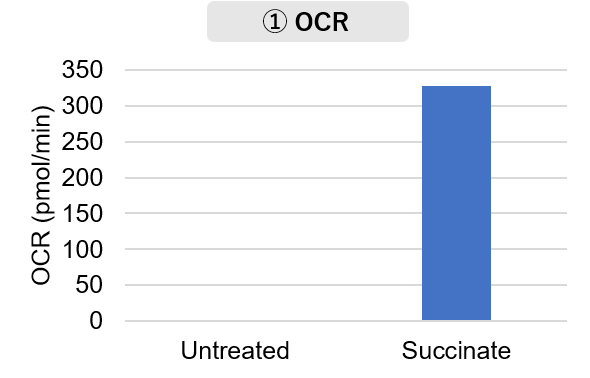
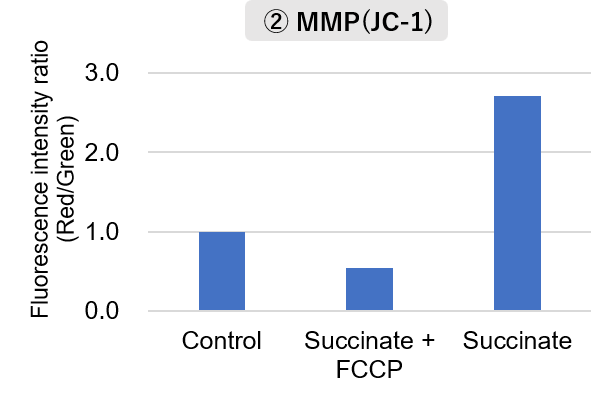
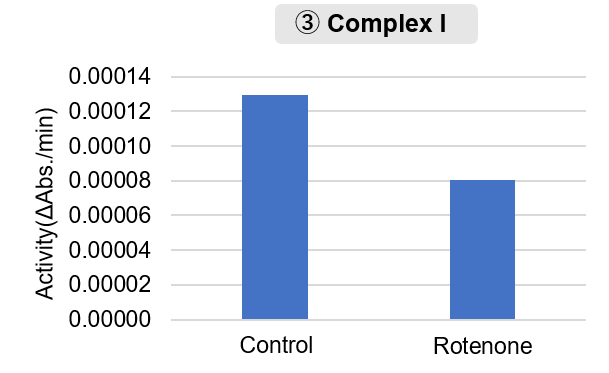
Mitochondria were isolated from mouse brain tissue, and oxygen consumption rate (OCR), mitochondrial membrane potential (MMP), and Complex I activity were measured.
The results showed that the addition of succinate, a substrate that activates Complex II of the electron transport chain, increased both OCR and MMP. In contrast, FCCP treatment reduced MMP, indicating that intact mitochondria were successfully fractionated.
Furthermore, in the Complex I activity assay, a decrease in activity was observed following treatment with rotenone, a Complex I inhibitor.
|
   |
<Product used>
Mitochondrial Fractionation: IntactMito Fractionation Kit for Tissue (Code: MT17)
OCR measurement: Extracellular OCR Plate Assay Kit (Code: E297)
MMP detection: JC-1 MitoMP Detection Kit (Code: MT09)
Complex I activity assay: MitoComplex- I Activity Assay Kit (Code: MT18) |
<Experimental Conditions>
OCR Measurement
Amount of mitochondria: 50 μg/well (as protein levels)
Succinate: 10 mmol/l
MMP Detection
Amount of mitochondria: 50 μg/well (as protein levels)
Succinate: 10 mmol/l, FCCP: 4 μmol/l
Complex I Activity Assay
Amount of mitochondria: 20 μg/well (as protein levels)
Rotenone: 10 μmol/l
|
|
| Metabolic Activity Assays
|