 Hidden sections will not be printed.
Hidden sections will not be printed.General Information
Nicotinamide adenine dinucleotide phosphate (NADP) is an important cofactor involved in reactions in the pentose phosphate pathway that is one of the metabolic pathways in cells. NADP exists as an oxidized form NADP+ and a reduced form NADPH in cells. NADPH is important for not only biosynthesis of fatty acids and cholesterol but also generation of reduced glutathione (GSH). In addition, recent study suggests that NADP+/NADPH associate with the extension of life span by carbohydrate restriction1).
NADP/NADPH Assay Kit-WST enables quantitation of the amount of total NADP+/NADPH, NADPH and NADP+ in cells and measurement of their ratio. The intracellular NADPH levels can be quantitated selectively by heat treatment of cell lysate with the extraction buffer in this kit. The intracellular NADP+ levels can also be determined by subtracting the amount of NADPH assayed from the amount of total NADP+/NADPH.
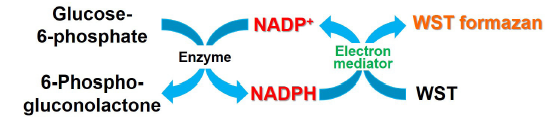
Fig. 1 Principle of NADP measurement by NADP/NADPH Assay Kit-WST
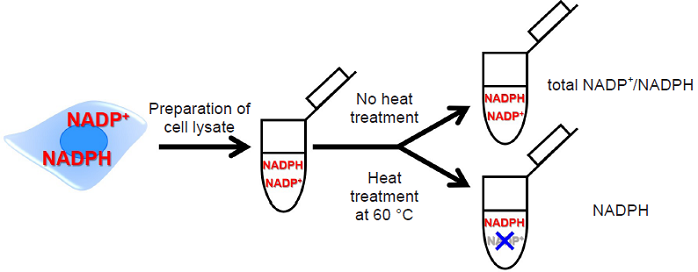
Fig. 2 Detection method of total NADP+/NADPH, NADPH and NADP+
Kit Contents
-
NADP/NADPH Extraction Buffer 20 ml x 1 NADP/NADPH Control Buffer 20 ml x 1 Standard Buffer 10 ml x 1 Assay Buffer 5.5 ml x 1 Dye Mixture (Red cap) x 1 Enzyme (Green cap) 110 μl x 1 Standard (Blue cap) x 1 Filtration Tube x 12
-
- The filtration tubes (12×) are sufficient for measuring 12 test samples.
For additional samples, commercially available filtration tubes (e.g., Nanosep Centrifugal Devices (10K)([OD010C33], PALL)) is recommended.
- The filtration tubes (12×) are sufficient for measuring 12 test samples.
Storage Condition
Store at 0-5 °C
Required Equipment
- Microplate reader (450 nm filter)
- 96-well microplate
- Incubator (37 °C, 60 °C)
- 20-200 μL multichannel pipette
- 100-1000 μL, 20-200 μL, 2-20 μL micropipettes
Precautions
- Equilibrate the kit to room temperature prior to use.
- Centrifuge the tube (Dye Mixture and Standard) briefly before opening the cap because the content may be on the tube wall or on the ceiling of the cap.
- Pipette the Enzyme before use to obtain the homogenous mixture since an enzyme is suspended in a liquid.
- Analyzing samples and standards in triplicate is recommended for accuracy.
- Since the enzymatic reaction starts immediately after the addition of Working solution to a well, use a multichannel pipette to minimize the experimental error from time lag in pipetting.
- Please prepare samples with different dilution rate and determine a suitable dilution rate to be ranging from 0 to 1 μmol/L.
Preparation of Solutions
Preparation of Dye Mixture stock solution
Add 550 μL of double-deionized H2O (ddH2O) to a Dye Mixture tube and dissolve it completely
- Centrifuge the tube briefly before opening the cap because the content may be on the tube wall or on the ceiling of the cap.
- Store the Dye Mixture stock solution at -20 °C with protection from light. The solution is stable at -20 °C for 2 months.
Preparation of Standard stock solution (10 mmol/L)
Add 20 μL of ddH2O to a tube of Standard and dissolve it with pipetting.
- Centrifuge the tube briefly before opening the cap because the content may be on the tube wall or on the ceiling of the cap.
- Standard stock solution should be kept in ice bath during your experiment.
- Store the Standard stock solution at -20 °C. The solution is stable at -20 °C for 2 months.
Preparation of Working solution
- Add Dye Mixture stock solution to a conical tube and dilute it with Assay Buffer.
- Add Enzyme to the solution prepared in step 1.
- Refer to the Table 1.
- Working solution is light-sensitive. Prepare the solution just before use and protect it from light by covering with aluminum foil. Please use up Working solution within that day.
| for 48 well | for 96 well | |
| Dye Mixture stock solution | 270 µL | 540 µL |
| Assay Buffer | 2.43 mL | 4.86 mL |
| Enzyme | 54 µL | 108 µL |
Table. 1 Examples of preparation of Working solution
General Protocol
1. Sample preparation
- Prepare a cell suspension (5-40×105 cells) in a 1.5 mL microtube.
- Centrifuge at 300×g for 5 minutes and remove the supernatant.
- Add 500 μL of PBS to each tube, suspend by pipetting, centrifuge at 300×g for 5 minutes and remove the supernatant.
- Add 300 μL of NADP/NADPH Extraction Buffer to each tube, lyse the cells by pipetting and centrifuge at 12,000×g for 5 minutes.
- Suction and discharge of the lysate using a syringe with the 25G needle (20-30 times) may work for hard-to-filter samples (e.g.,viscous) to be filtered smoothly in the downstream centrifugation.
- Transfer 250 μL of the supernatant to MWCO 10K filtration tube and centrifuge at 12,000×g for 10 minutes.
- At least 100 μL of sample is required for each total NADP+/NADPH and NADPH measurement (Total sample requirement: 200 μL).
- Extend the centrifugation time in case the filtrate is less than 200 μL.
- Transfer 100 μL of the filtrate into two 1.5 mL microtubes and use them as total NADP+/NADPH and NADPH sample.
- Incubate the sample solution for measuring the amount of NADPH at 60 °C for 60 minutes.
- This step is for decomposing NADP+ contained in the sample solution.
- Keep the sample solution for measuring the amount of total NADP+/NADPH on ice until assaying.
- After the incubation, cool the sample solution to room temperature.
- Add 100 μL of NADP/NADPH Control Buffer to each 1.5 mL microtube containing the sample solution for measuring the amount of total NADP+/NADPH and NADPH of Step 6. and Step 8. and use these solutions for measuring(Sample).
- 50 μL of sample per well is required.
- Please prepare samples with different dilution rate and determine the suitable dilution rate to be ranging from 0-1 μmol/L. Use NADP/NADPH Control Buffer for dilution.
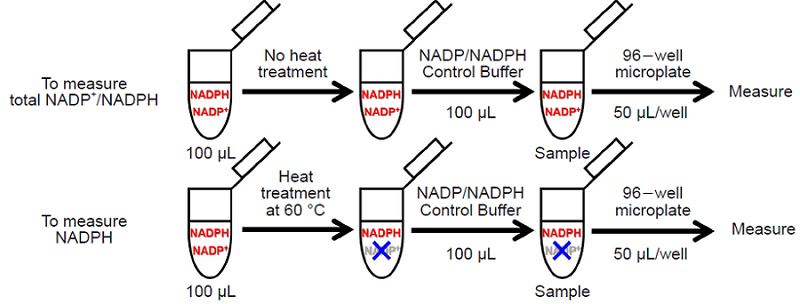
Fig. 3 Schematic diagram of the sample preparation protocol for measuring
total NADP+/NADPH and NADPH individually
2. Preparation of Standard solution
- Mix 2 μL of 10 mmol/L Standard stock solution and 198 μL of ddH2O in a microtube to prepare a 100 μmol/L Standard solution.
- Mix 5 μL of 100 μmol/L Standard solution and 495 μL of Standard Buffer in a microtube to prepare a 1 μmol/L Standard solution. Prepare Standard solutions (1, 0.5, 0.25, 0.125, 0.0625, 0.0313, 0.0157 and 0 μmol/L) as follows by serial dilution with Standard Buffer (Fig. 4).
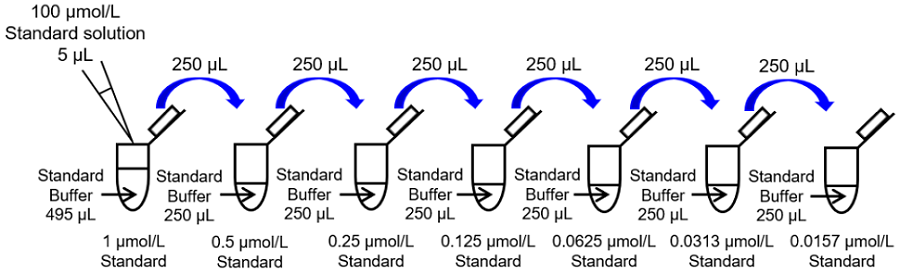 Fig. 4 Preparation of Standard solution
Fig. 4 Preparation of Standard solution
3. Measurement
- Add 50 μL of Standard solution and sample solutions to each well (Fig. 5).
- In order to obtain accurate data, we recommend triplicate measurement per sample.
- Add 50 μL of Working solution to each well.
- Since the enzymatic reaction starts immediately after the addition of Working solution to the well, use a multichannel pipette to minimize the experimental error from time lag in pipetting.
- Incubate the microplate at 37 °C for 60 minutes.
- Use a seal for the microplate to prevent evaporation of the solution during the incubation.
- Measure the absorbance at 450 nm by using a microplate reader.
- Determine the amount of total NADP+/NADPH and the amount of NADPH in the sample using a calibration curve.
- If the original samples were diluted for this assay, multiply the dilution rate with the determined value.
- The amount of NADP+ is calculated using the following equation from the amount of total NADP+/NADPH and NADPH.NADP+=total NADP+/NADPH-NADPH
-
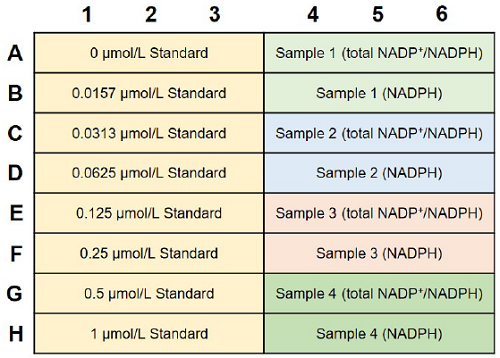
Fig. 5 An example of plate arrangement (n=3)
-
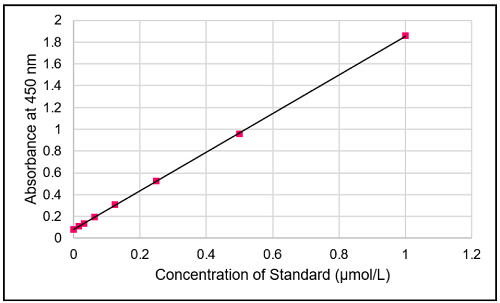
Fig. 6 Typical calibration curve of standard
Experimental Example
Analysis of total NADP+/NADPH, NADP+, NADPH and NADP+/NADPH ratio in Jurkat cells
- Jurkat cell suspensions (1.0 and 2.0×106 cells/tube) were prepared in a 1.5 mL microtube.
- The supernatants were removed after centrifugation at 300×g for 5 minutes.
- PBS (500 μL) was added to each tube, and the supernatants were removed after centrifugation of the tubes at 300×g for 5 minutes.
- NADP/NADPH Extraction Buffer (300 μL) was added to each tube and the cells were lysed by pipetting, and then the solutions were centrifuged at 12,000×g for 5 minutes.
- The supernatant (250 μL) was transferred to MWCO 10K filtration tube, and centrifuged at 12,000×g for 10 minutes.
- The filtrate was divided into two 1.5 mL microtubes by 100 μL, and used as the sample solution for the amount of total NADP+/NADPH and NADPH. The former sample solution was kept on ice until assaying.
- The sample solution for the amount of NADPH was incubated at 60 °C for 60 minutes. After the incubation, the sample solution was cooled to room temperature.
- NADP/NADPH Control Buffer (100 μL) was added to each 1.5 mL microtube containing the sample solution for measuring the amount of total NADP+/NADPH and NADPH, and these solutions were used for measuring.
- The sample solution (50 μL) and Standard solution (50 μL) were added to each well.
- Working solution (50 μL) was added to each well.
- he 96-well microplate was incubated at 37 °C for 60 minutes.
- The absorbance at 450 nm was measured by using a microplate reader, and the amount of total NADP+/NADPH and NADPH in the sample using a calibration curve. The amount of NADP+ was calculated by subtracting the amount of NADPH from the amount of total NADP+/NADPH.
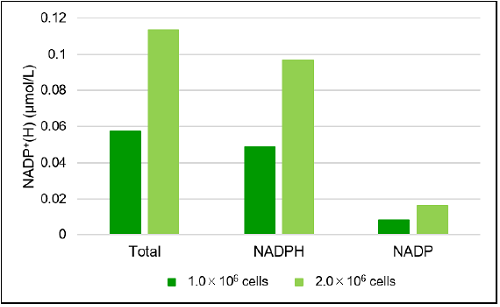
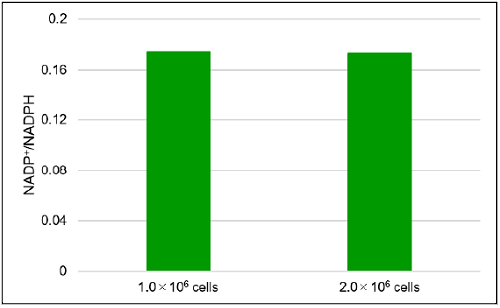
Fig. 7 The amount of total NADP+/NADPH, NADP+, NADPH and NADP+/NADPH ratio in Jurkat cells
Reference
- Richard L. Veech, et al., IUBMB Life., 2017, 69, 305.
Frequently Asked Questions / Reference



N510: NADP/NADPH Assay Kit-WST
Revised May., 23, 2023