 Hidden sections will not be printed.
Hidden sections will not be printed.General Information
Ferroptosis is a type of cell death caused by the abnormal accumulation of lipid peroxides in an iron‑dependent manner. During the process of lipid peroxidation, lipid radicals are generated through the oxidation of lipids. Therefore, to accurately evaluate the occurrence of ferroptosis, it is important to detect not only lipid oxidation and lipid peroxides but also lipid radicals1).
NBD-Pen is a fluorescent dye that selectively reacts with intracellular lipid radicals. When added to cultured cells, NBD-Pen passes through the cell membrane and emits a fluorescent signal upon reaction with lipid radicals. Unlike Lyso-NBD-Pen (Product Code: L271), NBD-Pen does not localize to a specific organelle, allowing detection of lipid radicals throughout the cell.
Contents
| Lipid Radical Probe -NBD-Pen- | 10 nmol × 1 |
| Lipid Radical Probe -NBD-Pen- | 10 nmol × 5 |
Storage Condition
Store in a cool and dark place.
Required Equipment and Materials
- Fluorescence microscope, or flow cytometer
- Incubator (37℃)
- Micropipettes (100–1000 µl, 20–200 µl, 1–10 µl)
- Microtubes
- Medium or Hanks' Balanced Salt Solution (HBSS)
Precaution
- Centrifuge the tube briefly before opening the cap because the contents may adhere to the tube wall or the inside of the cap.
- Please refer to Table 1 for suitable fluorescence wavelengths for each application.
Table 1. Recommended filter settings for NBD-Pen
| Applications | Fluorescence microscope | Flow cytometer |
| Measurement wavelength |
・Confocal microscope Ex/Em: 488/490–600 nm ・Fluorescence microscope GFP filter |
FITC filter |
Preparation of Solutions
Preparation of 1 mmol/l NBD-Pen DMSO stock solution
Add 10 μl of DMSO to a tube containing 10 nmol of NBD-Pen and dissolve by pipetting to prepare a 1 mmol/l NBD-Pen DMSO stock solution.
- The 1mmol/l NBD-Pen DMSO stock solution is stable at −20℃ for 1 month.
Preparation of 1 µmol/l NBD-Pen working solution
Dilute the 1 mmol/l NBD-Pen DMSO stock solution 1:1000 in medium to prepare 1 µmol/l NBD-Pen working solution.
- The working solution cannot be stored and must be prepared each day freshly.
- Refer to Table 2 for the amount of working solution required by vessel type.
Table 2. Required amount of the working solution by vessel type
| Vessel | 35-mm dish | ibidi 8-well plate | 96-well black plate (clear bottom) |
| Appropriate amount | 2 ml | 200 μl/well | 100 μl/well |
General Protocol
- Seed cells in a vessel. Culture the cells at 37°C overnight in a 5% CO2 incubator.
- Discard the supernatant and wash the cells once with medium.
- Discard the supernatant, add an appropriate volume of 1 µmol/l NBD-Pen working solution to the vessel, and incubate at 37°C for 30 min in a 5% CO2 incubator.
- Discard the supernatant and wash the cells twice with medium.
- Add medium containing ferroptosis inducers or inhibitors, and incubate the cells at 37°C in a 5% CO2 incubator for an appropriate time.
- Discard the supernatant and wash the cells twice with HBSS.
- Observe the cells under a fluorescence microscope or measure fluorescence signals using a flow cytometer.
Usage Example 1
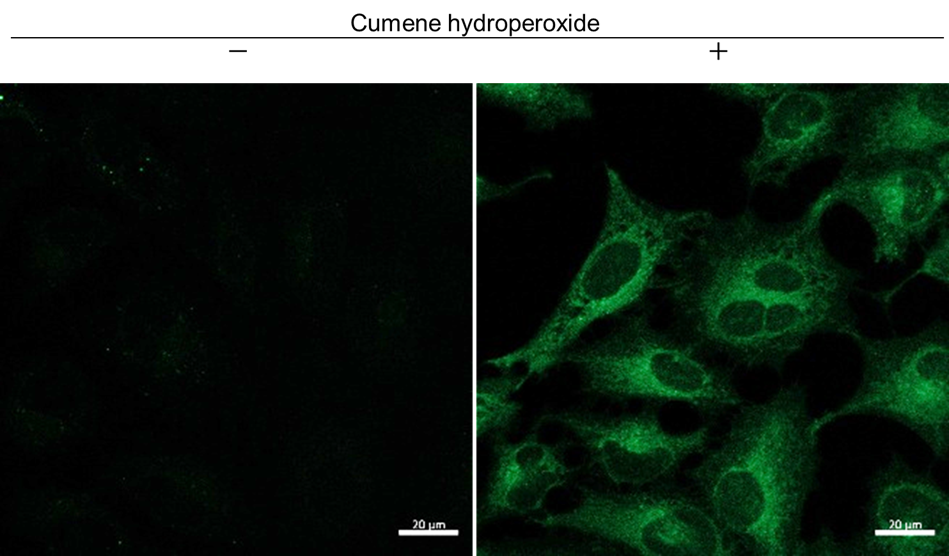
Detection of lipid radicals in HeLa cells treated with cumene hydroperoxide using a confocal laser microscope
- HeLa cells (3×104 cells/well) in MEM (supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin) were seeded in an ibidi 8-well plate and incubated at 37°C overnight in a 5% CO2 incubator.
- After the supernatant was removed, the cells were washed once with medium. Then, 1 µmol/l NBD-Pen working solution (200 µl/well) was added to the cells, and they were incubated at 37°C for 30 min in a 5% CO2 incubator.
- The supernatant was removed, and the cells were washed twice with HBSS.
- Cumene hydroperoxide (200 µmol/l) dissolved in HBSS was added to the cells, and they were incubated at 37°C for 1 h in a 5% CO2 incubator.
- The supernatant was removed, and the cells were washed twice with HBSS. The wells were refilled with HBSS.
- The cells were observed under a confocal laser microscope.
 |
Detection: Confocal laser microscope |
Figure 1. Fluorescence images of HeLa cells obtained with a confocal laser microscope
Usage Example 2
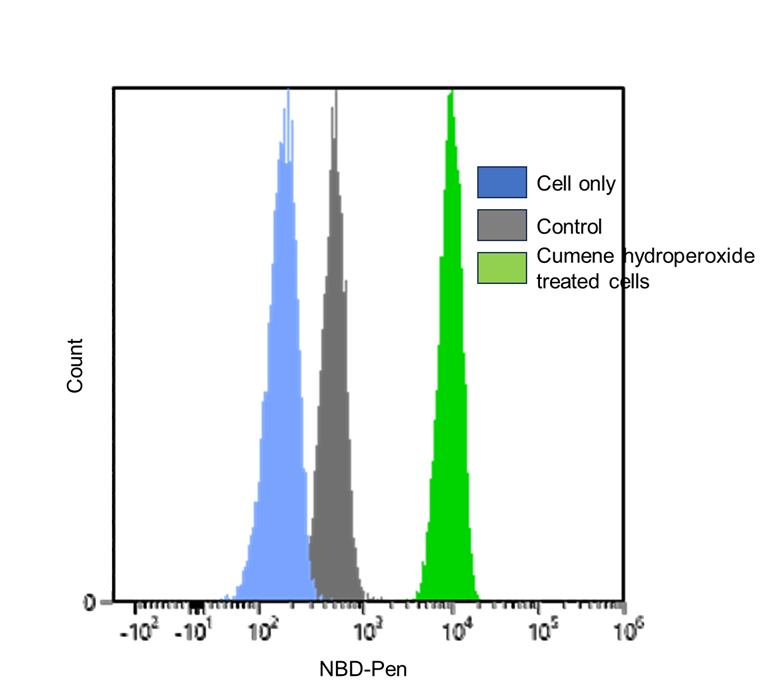
Detection of lipid radicals in HeLa cells treated with cumene hydroperoxide using a flow cytometer
- HeLa cells (3×105 cells/well) in MEM (supplemented with 10% fetal bovine serum and 1% penicillin–streptomycin) were seeded in a 6-well plate and incubated at 37°C overnight in a 5% CO2 incubator.
- After the supernatant was removed, the cells were washed once with medium. Then, 1 µmol/l NBD-Pen working solution (2 ml/well) was added to the cells, and the cells were incubated at 37°C for 30 min in a 5% CO2 incubator.
- The supernatant was removed, and the cells were washed twice with medium.
- Cumene hydroperoxide (200 µmol/l) dissolved in HBSS was added to the cells, and they were incubated at 37°C for 1 h in a 5% CO2 incubator.
- After removing the supernatant, the cells were washed once with HBSS and detached from the plate using 0.25% trypsin–EDTA.
- The detached cells were collected into a 1.5‑ml tube containing serum-supplemented medium and centrifuged at 300 × g for 5 min.
- The supernatant was discarded, 1 ml of PBS was added to resuspend the cells, and the cells were centrifuged at 300 × g for 5 min.
- The supernatant was discarded, and the cells were resuspended in 1 ml of PBS.
- The samples were passed through a cell strainer for flow cytometry and analyzed using a flow cytometer.
 |
Detection: Flow cytometer |
Figure 2. Fluorescence signals from HeLa cells, detected using a flow cytometer
Reference
1) K. Yamada et al., Nature Communications, 2025, 16, 2554.
Frequently Asked Questions / Reference



L272: Lipid Radical Probe -NBD-Pen-
Revised Apr., 13, 2026